- Products
- Next Generation Sequencing Kits
- Clinic Laboratory
- Laboratory equipment
- Pathology and anatomy
- Analytical & test instruments
- Diagnostic kits
- Rapid Diagnostic
- Reagents and chemicals
- IVF Products
- Microbiology
- Laboratory furniture
- Kits for Veterinary Medicine
- Laboratory consumable
- Promotions
- News
- Service
- About Us
- Careers
- Training
- Contacts
- Product News
- HiSeq 3000/HiSeq 4000

New horizons in effective prevention and acurate diagnostic of Cystic Fibrosis
Муковисцидоза (Cystic Fibrosis – CF) e най-честото разпространено, тежко, практически нелечимо генетично заболяване сред бялата раса. Честотата на тежките форми на болестта у нас е около 1: 2500 новородени. Всеки десети случай при „мъжки фактор” на безплодие с установен нисък брой на сперматозоиди се дължи на дефект в гена за муковисцидоза. Като се прибавят, късните форми на проява и се вземе предвид продължителността и стойността на поддържащото лечение, заболяването може да се определи категорично като „социално значимо”.
Муковисцидозата се причинява от мутации в гена – CFTR. Този ген кодира информация за синтеза на белтък (протеин) – Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), който изпълнява роля на канал в мембраната на всяка клетка като засяга предимно жлезите с външна секреция, произвеждащи: мукус, пот, слюнка, сълзи и някои храносмилателни ензими. Генът се намира в дългото рамо на Хромозома 7. Нормалното функциониране на белтъка позволява оптимален транспорт на хлорните йони, които регулират придвижването на вода в тъканите. Така се образува тънък слой мукус. Последният е с решаващо значение за нормалното функциониране на белият дроб, както и на други органи и системи, като: половата система при мъжа, панкреас, черния дроб, жлъчката, кожата и др.
За да се прояви заболяването при даден индивид той трябва да носи по един мутантен ген от родителите си. При двама носители рискът за поява на болен в поколението е много висок – 25%. Този модел на унаследяване се определя като автозомно рецесивен. В случай, че само едното копие на гена е с мутация (независимо дали е унаследено от майката или от бащата) заболяването не се проявява, въпреки, че някои последни проучвания свързват проявата на безплодие у мъжа с дефекти само на едното копие на CFTR гена. Честотата на носителство на муковисцидоза у нас е около 1:30.
След проекта за разчитане (секвениране) на човешкия геном, приключил през 2003 г. CFTR гена е напълно разчетен – знае се точното подреждане в него на 4-те бази на ДНК и извънгенните контролни региони. Установени са над 1600 различни мутации и съответно вариации (алели) на гена. Много от тях водят до тежки форми на болестта. Най-честите мутации на гена, както и пълният набор от такива могат да се намерят в изградена база данни на www.CFTR2.org
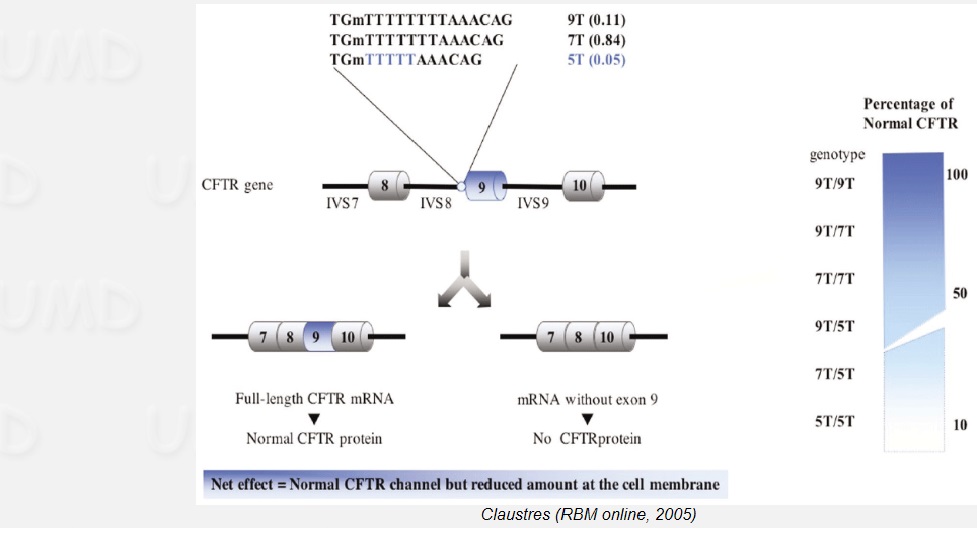
В момента капилярното секвениране продължава да бъде „златен” стандарт за откриване и потвърждаване на мутации в гена за муковисцидоза. То е скъп и трудоемък процес, поради което е неприложимо за включването му в масови скринингови програми. Това е особено изразено при генетично разнообразни популации, каквито са българите. За разлика от ромите, баските и датчаните, където обикновено се намира само един вид мутация, при българите са открити над 35 различни мутации, като при 8-10% от клинично проявените форми, мутациите все още не са доказани (www.lmpbg.org).
След въвеждане на Новото поколение секвенатори, с които разполага не само Центъръ по молекулна медицина към Медицински Университет – София (www.mmcbg.org) , установяването на широк набор от различни мутации на гена, вече е напълно възможен и то в десетки проби едновременно (48бр.).


Това намалява значително цената и прави по-достъпно генетично изследване за носителство на CFTR генни мутации.
Диагностиката на муковисцзидозата и носителство на дефектен CFTR ген обикновено се установява късно. Най-често между 4 -5 годишна възраст с изяви на клиничните прояви на болестта – чести и не повлияващи се от обичайните комбинации на антибиотици възпаления на белите дробове и т.н. Тук не се включват „леките” форми, които се появяват след 40-50 годишна възраст и тези, които водят до мъжки стерилитет.
За първи път сега, с въвеждането на новите геномни технологии, е възможно установяването на заболяването още в първите 2- 3 дни от раждането на детето. Нещо повече, става възможно масовото доказване на носителство преди и поставяне на диагноза по време на бременността.
Важно е да се отбележи, че за първи път до сега, може да се осъществи цялостно директно генетично изследване на:
• Новородени, деца и възрастни с клинични данни за муковисцидоза и положителен потен тест;
• родственици с доказано заболяване на един член от фамилията с оглед да се определи кои са носителите (асимптомни) на това тежко хронично и социално значимо заболяване в рода с оглед последваща превенция.
• бъдещи майки (независимо от липсата на данни за болни родственици) с цел доказване на носителство и в случай на доказано нисителство в двамата пяртньора предлагане на дородова диагностика в 11-20 седмица на бременността.
• при мъже с нисък брой на сперматозоидите.
С достиженията на съвременната репродуктивна медицина и метода Ин Витро, може да се избере ембрион който не е носител на тежък генен дефект в случай, че и двамата кандидат родители или донори са носители на мутации в CFTR гена.С достиженията на съвременната репродуктивна медицина и метода Ин Витро, може да се избере ембрион който не е носител на тежък генен дефект в случай, че и двамата кандидат родители или донори са носители на мутации в CFTR гена.
Конкретно самото изследване се състои в изолиране на ДНК за 48 пациенти едновременно в един пуск на апарата МайСик. Изолирането на ДНК се подготвя за анализ и деректно секвениране чрез комплекта реактиви Cystic Fibrosis Carrier Screening Assays.


Тестът е предназначен за масово изследване на новородени с цел ранна диагностика и по-ефективно лечение. Той открива рекорден брой -162 болествотворни варианти (мутации) на CFTR гена. Това е особено ефективно при популации с множество редки мутации. За сравнение, всички до сега известни теста откриват не повече от 36 варианта на гена, като не включват част от мутациите характерни за Балканския регион. Предлаганият тест е с поне 5 пъти по-големи възможности и откриваемост над 99% от известните мутации.
Този тест определя варианти на изследвания ген -162 грижливо подбрани от световни експерти в тази област от базата данни CFTR2 за рекордно обхващане на различни демографски области.

Този нов тест подобрява в пъти диагностиката на заболяването. С досега използваните в световен мащаб тестове за скрининг чрез директен генетичен анализ се установяват огромен обем фалшиво отрицателни резултати поради ограниченото откриване на възможните мутации.
Подобряването на диагностиката, ранното откриване в неонаталният период, възможността за откриване на рискови семейства и провеждане на дородова диагностика ще доведе до значително:
(1) ограничаване на заболеваемостта;
(2) намаляване на усложненията при муковисцидоза, проявяваща се в по-късен период от развитието на индивида,
(3) повишаване на успеваемостта при ин витро в случаите с „мъжки фактор”.
В крайна сметка ще се балансират разходите и ще се създаде допълнителна възможност за насочване на освободените при профилактиката средства към подобряване качеството на живот на страдащите от муковисцидоза.
С прилагането на теста като рутинен и скринингов метод в неонаталния период ще се покачи рязко качеството на детското здравеопазване, повишаване на преживяемостта на засегнатите, а от там и положителен отзвук на обществото в лицето на неправителствените организации и сдружения на засегнатите от това заболяване, пациентски организации и т.н.